| ** | Latin American Journal of Clinical Sciences and Medical Technology is an open access magazine. To read all published articles and materials you just need to register Registration is free of charge. Register now If you already have registered please Log In | ** |




aDepartamento de Inmunología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Ciudad de México, México; bCONACYT-Facultad de Odontología, Universidad Autónoma Benito Juárez de Oaxaca, Ciudad Universitaria, Oaxaca de Juárez, Oaxaca, México; cUnidad de Investigación Médica en Trombosis, Hemostasia y Aterogénesis, Instituto Mexicano del Seguro Social, Ciudad de México, México.
Autor para correspondencia: , . Números telefónicos: ; e-mail: amajlufc@gmail.com

Lat Am J Clin Sci Med Technol. 2020 Sep;2:197-207.
Recibido: 06 de agosto, 2020
Aceptado: 11 de septiembre, 2020
Publicado: 29 de septiembre, 2020
Vistas: 1176
Descargas: 10

Introducción. La hemofilia A (HA) es una enfermedad genética, recesiva y ligada al cromosoma X en el cual se codifica el factor hemostático VIII (FVIII). Desde hace años, el tratamiento de la HA se realiza restituyendo el factor deficiente al paciente. La complicación más temida asociada al tratamiento es la generación de aloanticuerpos con efecto neutralizante sobre el FVIII, los cuales impiden la función de dicho factor. Material y métodos. Revisión narrativa selectiva en PubMed de los términos relacionados con inhibidores del FVIII de artículos escritos en inglés o español, entre los años 2000 y 2020. Resultados. Para generar esta revisión incluimos 59 artículos originales y de revisión relacionados con la generación de inhibidores en HA. Discusión. Cuando el paciente inicia tratamiento también comienza la exposición a una proteína cuyo sistema inmune desconoce, por lo que se generan inhibidores dirigidos contra el FVIII. Los inhibidores afectan la hemostasia, impiden la unión del FVIII a otros factores hemostáticos, inhiben la activación del FVIII, evitan la liberación del FVIII del complejo VWF-FVIII o pueden aumentar la proteolisis del FVIII. El desarrollo de inhibidor depende de interacciones entre factores relacionados con el paciente y el tratamiento. La respuesta anti-FVIII es T dependiente y el bazo es preponderante. La generación de inhibidores puede explicarse por las teorías del daño y de lo propio y no propio. Los factores de riesgo de inhibidores asociados al paciente (mutaciones del F8 y haplotipo del FVIII) podrían relacionarse con la última; los asociados al tratamiento (dosis y el origen de FVIII) se relacionarían con la primera.
Introduction. Hemophilia A (HA) is a recessive genetic disease linked to the X chromosome, in which the hemostatic factor VIII (FVIII) is codified. For years, the HA treatment consists of restoring the deficit of such a factor to the patient. The most widely feared complication associated with the treatment is the production of alloantibodies with a neutralizing effect on the FVIII, which inhibits the performance of the factor. Material and Methods. Selective narrative review in PubMed of the terms related to the FVIII inhibitors in articles written in English or Spanish, published between 2000 and 2020. Results. To carry out this review, we included 59 original and review articles related to the production of HA inhibitors. Discussion. When the patient starts treatment, the immune system is also beginning to be exposed to an unknown protein. Thus, the production of inhibitors directed against the FVIII starts. The inhibitors affect hemostasis, interfere with union of FVIII to other hemostatic factors, inhibit FVIII activation, block the release the FVIII from the VWF-FVIII complex, or they may rise FVIII prpoteolysis. Inhibitor development depends on the factors related to the patient and the treatment. The anti-VIII response is T-dependent, and the spleen is preponderant. The production of inhibitors may be explained by the danger model and self and non-self. The risk factors of the inhibitors associated with the patient (mutations in F8 and FVIII haplotype) might be related to the last one; the ones pertaining to treatment (doses and the FVIII origin) are associated with the second one.
La hemofilia A (HA) es una enfermedad genética, recesiva y ligada al cromosoma X en el cual se codifica el factor hemostático VIII (FVIII). Algunas alteraciones estructurales de este gen condicionan una deficiencia cuantitativa o funcional del FVIII en la HA. Los factores VIII y IX tienen un papel hemostático fundamental. La generación de trombina es un evento biológico toral porque esta enzima es parte del complejo molecular que forma el coágulo luego de una lesión vascular. El FVIII acelera miles de veces la velocidad de las reacciones, las cuales generan trombina. El FIXa (la enzima) se une al FVIIIa (la coenzima) en un ambiente lipídico para formar el complejo «diezasa intrínseca», el cual genera trombina con una eficiencia 106 veces mayor que la del FIX aislado. Por lo tanto, la deficiencia de estos dos factores se traduce en hemofilia.
Desde hace años, el tratamiento de la HA se realiza restituyendo el factor deficiente al paciente. El factor era de origen plasmático, con los riesgos consecuentes por su origen biológico, esencialmente la transmisión de infecciones. Sin embargo, superadas las epidemias de los virus de inmunodeficiencia humana y hepatitis C, la complicación más temida asociada al tratamiento de HA es la generación de aloanticuerpos con efecto neutralizante sobre el FVIII. Al aloanticuerpo que impide la función del FVIII lo llamamos «inhibidor», el cual neutraliza o impide la función o aumenta la degradación del FVIII. Los inhibidores afectan hasta 30% de los pacientes con HA.1 El inhibidor dificulta el tratamiento ya que el control de la hemorragia es cada vez más difícil, lo cual ensombrece la esperanza y calidad de vida del paciente, además de que los costos económicos se multiplican significativamente.
Debido a la importancia que hoy tienen los inhibidores sobre la atención y los costos (sociales, familiares y económicos) de la HA, es necesario revisar cómo influyen en su generación tanto las explicaciones clásicas (teoría de lo propio y lo no propio), como los conceptos generados a partir de las teorías nuevas (teoría del daño). Por lo tanto, el objetivo de esta revisión es hacer un análisis conciso, pero eficiente, de las teorías que explican la generación de inhibidores asociados a la HA.
Realizamos una revisión narrativa selectiva en PubMed de artículos escritos en inglés o español, publicados entre los años 2000 y 2020. Para tal efecto, realizamos una búsqueda de los artículos que comprendieran los términos «hemophilia A», «plasma derived factor VIII», «recombinant FVIII», «FVIII inhibitors», «bleeding», «hemophilia A treatment», «FVIII immunogenicity», y «FVIII alloantibodies». Analizamos la evidencia disponible para poner en contexto el problema de los inhibidores en HA y la que atañe a las explicaciones inmunológicas para la aparición de los inhibidores. Seleccionamos la información que, a nuestro juicio, era de relevancia para esta revisión. Aunque la evidencia publicada con respecto al tema desarrollado es extraordinariamente amplia, intentamos dirigir la selección de trabajos hacia las dos teorías más relevantes en este momento: la teoría de lo propio y lo no propio y la teoría del daño, intentando tener los menos sesgos posibles. Al final, se analizaron más de 238 artículos relacionados con el tema propuesto. Sin embargo, incluimos finalmente 59 artículos originales y de revisión relacionados francamente con la explicación de la aparición de inhibidores en HA.
FVIII
El FVIII es una coenzima glicoproteica de la fase líquida de la hemostasia (antes conocida como «cascadas de coagulación»). Su expresión insuficiente o disfuncional causa la enfermedad conocida como HA. Como ya mencionamos, esta enfermedad genética es recesiva y ligada al cromosoma X; es heredada en el 70% de los casos y en el 30% restante es consecuencia de una mutación de novo.2,3
El gen del FVIII se localiza en la punta del brazo largo del cromosoma X, abarca 180 kb y comprende 26 exones que dan como producto final una proteína madura de 2,332 aminoácidos. El FVIII tiene una estructura molecular A1-a1-A2-a2-B-a3-A3-C1-C2 en su forma monocatenaria, en donde A1, A2, B, A3, C1 y C2 son los dominios de la proteína mientras que a1, a2 y a3 son regiones ácidas ricas en tirosinas sulfatadas. Debido al procesamiento postraduccional, el FVIII se secreta como proteína heterodimérica que contiene una cadena pesada (A1-a1-A2-a2-B) unida por un ion metálico (Mn2+ y/o Ca2+) a una cadena ligera (a3-A3-C1-C2). La unión se encuentra entre los dominios A1 y A3.2,4,5
El sitio donde se produce predominantemente el FVIII son las células endoteliales sinusoidales hepáticas6; sin embargo, el mRNA del FVIII está presente en otros órganos como bazo, pulmón y riñones, los cuales pudieran contribuir a los niveles circulantes de este factor.4
El factor von Willebrand (VWF) y el FVIII circulan en sangre como un complejo no covalente que regula la agregación plaquetaria y la formación del coágulo de fibrina. El VWF se requiere para la adhesión plaquetaria al subendotelio y la supervivencia del FVIII circulante. El complejo FVIII-VWF circulante se elimina principalmente al interactuar con la proteína relacionada con el receptor de lipoproteínas de baja densidad (LRP1), localizada en la membrana hepatocítica. Los dominios C3, A3 y A2 del FVIII interactúan con el LRP1 para eliminarse eficientemente. La presencia de múltiples sitios para la interacción FVIII-LRP asegura la eliminación de cadenas no unidas y del dominio A2 del torrente sanguíneo.7
Hemofilia A
La HA es consecuencia de una mutación cuyo propositus la heredará con el patrón recesivo ligado a X.8 Dado que los dos tipos de hemofilia tienen un patrón recesivo, clínicamente se manifiestan casi siempre sólo en varones; las mujeres son portadoras de la alteración genética y tienen sintomatología mínima, si bien, excepcionalmente, sufren hemorragias importantes bajo condiciones muy especiales.
En la HA, las alteraciones cromosómicas son, generalmente, mutaciones puntuales (46% de los casos), rearreglos (inversiones, 42%), deleciones (8%) y mutaciones no identificadas (4%). La prevalencia mundial aproximada es 1 caso/10,000 varones. Se calcula que en el mundo hay casi 400,000 personas con HA1 y en México se estiman casi 6,300 casos; la cifra exacta la desconocemos porque no contamos con un registro nacional ni diagnóstico epidemiológico preciso.
La HA limita todos los aspectos de la vida biológica, psicológica y social del enfermo; para el sistema de salud y la sociedad, a pesar de su prevalencia baja, su efecto es alto. Es oportuno resaltar que, mundialmente, sólo 30% de los pacientes recibe un tratamiento óptimo.9 Hasta el año 2020, en México esto no era diferente; hoy se tiene un sistema consolidado de tratamiento que se espera abarque a casi 100% de los casos.
Los puntos importantes para el diagnóstico son la historia clínica para buscar antecedentes familiares y el patrón de herencia, la semiología de la hemorragia y la exploración física. La HA se caracteriza por hemorragias que no aparecen inmediatamente después de una lesión vascular. En orden de frecuencia, estas hemorragias son hemartrosis (en articulaciones de carga: rodillas, tobillos y codos, principalmente), hematomas musculares profundos y hemorragias cerebrales. Éstas constituyen 95% de las hemorragias del paciente; sin embargo, los eventos hemorrágicos pueden afectar cualquier parte del cuerpo.
Los estudios de laboratorio son importantes, en especial, la cuantificación de la actividad funcional del FVIII ya que existe una relación entre tendencia hemorrágica y grado de deficiencia del factor, aunque existen excepciones.10 El nivel funcional del FVIII permite clasificar la HA en grave (<1% de la actividad del FVIII), moderada (entre 1 y 5%) y leve (entre 5 y 40%).
Hoy contamos con productos liofilizados que contienen FVIII de origen recombinante (rFVIII) u obtenido del plasma de donadores sanos (dpFVIII). El estándar de oro del tratamiento moderno de la HA es reponer el factor deficiente, preferentemente, aplicándolo de manera profiláctica o como tratamiento oportuno a demanda. Éste consiste en infundir el FVIII en dosis óptima en el menor tiempo posible luego de la lesión vascular. La profilaxis consiste en aplicar regularmente el FVIII para mantener concentraciones adecuadas en el enfermo por el mayor tiempo posible; es la primera opción para la Organización Mundial de la Salud (OMS).11-14 La profilaxis previene significativamente las hemorragias y el deterioro articular15 al permitir que el fenotipo del paciente pase de grave a moderado con lo cual se logra casi normalizar la actividad física, el rendimiento escolar y la integración social.12,16-18
La HA es una enfermedad devastadora que ha sido contenida con bastante éxito aunque con un costo económico muy elevado. Por ello, el tratamiento óptimo no está disponible en muchos países. Idealmente, todo paciente debiera iniciar tratamiento antes de los dos primeros años o luego de la primera hemorragia articular; obviamente, esto aumenta la exposición a una proteína que desconoce. La consecuencia es la generación de inhibidores dirigidos contra el FVIII que se está aplicando.
Inhibidores del FVIII
El desarrollo de aloanticuerpos neutralizantes del FVIII, o inhibidores, representa la complicación principal del tratamiento sustitutivo de la HA. La frecuencia de inhibidores en la HA grave es de 20-33% y de 3-13% en la HA leve o moderada.19,20
Además de los inhibidores, otra población de anticuerpos está presente en individuos sanos y en pacientes con HA sin ejercer actividad inhibitoria de la fase líquida de la hemostasia; están dirigidos contra epítopos no funcionales del FVIII y reciben el nombre de anticuerpos no neutralizantes (NNA, non-neutralizing antibodies). Su prevalencia aproximada es de 2-3% en sanos y de 12-54% en hemofílicos.17 Los inhibidores se cuantifican en unidades Bethesda (UB), donde 1 UB/ml inhibe 50% de la actividad del FVIII presente en la muestra.20
La respuesta inmune mediada por inhibidores del FVIII no está isotípicamente restringida e involucra todas las subclases de IgG. Sin embargo, en pacientes con HA congénita y adquirida los anticuerpos IgG1 e IgG4 son los que predominan. En cambio, las inmunoglobulinas IgG1 e IgG3 son las que predominan en pacientes sin inhibidores.21,22
Los inhibidores afectan la fase plasmática de la hemostasia por diferentes mecanismos:
- impiden estéricamente la unión del FVIII con otros factores hemostáticos,
- inhiben la activación del FVIII por trombina e
- inhiben la liberación del FVIII del complejo VWF-FVIII.
Asimismo, los inhibidores afectan la hemostasia al ejercer actividad proteolítica sobre el FVIII.19
Factores de riesgo para producir inhibidores del FVIII
El desarrollo de inhibidor depende de las interacciones entre múltiples factores relacionados con el paciente y el tratamiento (Tabla 1).
| Tabla 1. Factores de riesgo asociados al desarrollo de inhibidores del FVIII | |
|---|---|
| Asociados al paciente | Asociados al tratamiento |
| Mutación en el gen del F8 | Origen del producto (plasmático o recombinante) |
| Origen étnico | Régimen de tratamiento (profilaxis o a demanda) |
| Historial familiar de inhibidores | Dosis de FVIII administrado |
| Dosis basal de FVIII | Presencia de agregados |
| Haplotipo del FVIII | Relación entre dosis real y dosis mostrada en la etiqueta |
| Grupo sanguíneo | |
| Anticuerpos no neutralizantes | |
El factor de riesgo más importante atribuible al paciente para desarrollar inhibidor recae en las mutaciones del gen F8, donde las deleciones grandes, las mutaciones sin sentido y las que cambian el marco de lectura del gen imprimen el riesgo más grande, en comparación con mutaciones con sentido equivocado.23 Otro factor de riesgo genético es el haplotipo de FVIII tanto del paciente como del producto de reemplazo.
Se conocen seis haplotipos del FVIII (H1-H6), resultado de la combinación de cuatro polimorfismos no sinónimos de un solo nucleótido: G1679A, A2554G, C3951G, A6940G. Los haplotipos H3, H4 y H5 se relacionan con la presencia de epítopos inmunodominantes en el FVIII.24 La prevalencia de los haplotipos en general no cambia; el más prevalente es el H1, seguido del H2 y H3. La diferencia de haplotipo entre el producto de reemplazo y el FVIII endógeno del paciente se relaciona con aumento del riesgo de desarrollar inhibidor.24,25
El grupo sanguíneo AB0 también influye en el desarrollo de inhibidores. Se ha observado que 14.9% de los pacientes con HA grave y de origen caucásico desarrolla inhibidores si el grupo sanguíneo es 0; sin embargo, este porcentaje se eleva a 33.1% en pacientes de grupo sanguíneo no 0. Lo anterior puede deberse a que pacientes con grupo sanguíneo 0 tienen 25% menos VWF en la circulación, lo que posiblemente acorte la vida media del FVIII y el tiempo de exposición de este factor al sistema inmune.26
Además de los factores genéticos existen factores adquiridos. La actividad del FVIII es uno de ellos, es decir, si la actividad basal de FVIII es <0.01 UI/ml, el riesgo de desarrollar inhibidor se duplica vs. la actividad basal entre 0.01 y 0.02 UI/ml. No obstante, el riesgo aumenta tres veces en pacientes con historia familiar de inhibidor. Además, cuando el factor se administra a muy temprana la edad, el riesgo es mayor; hasta 41% de los pacientes con HA grave desarrolla inhibidores si reciben FVIII antes del primer mes de vida; el porcentaje disminuye a 30, 23, 20 y 18%, si la edad al inicio del tratamiento es entre 1 y 6 meses, entre 6 y 16 meses, entre el 12 y 18 meses y después de los 18 meses del nacimiento, respectivamente.27
Al igual que la historia familiar de inhibidor, la presencia de NNA favorece el desarrollo de inhibidores. Los pacientes con NNA tienen 1.78 veces más riesgo de desarrollar inhibidores que los enfermos sin NNA; sin embargo, el riesgo es 2.62 veces mayor cuando los pacientes con NNA desarrollan inhibidores de alta respuesta.28
De los factores de riesgo atribuibles al tratamiento, el origen del FVIII es de suma importancia. El FVIII proviene de dos fuentes, del plasma (pdFVIII) o recombinantes (rFVIII). El tratamiento con rFVIII genera más inhibidores que los pdFVIII.29 Entre las posibles explicaciones se encuentra la diferencia en el perfil de glicosilación entre los productos. Por ejemplo, los recombinantes tienen epítopos no humanos como el alfa gal (Gala1,3Gal) o el ácido N-glicolilneuramínico.30
Otra diferencia entre los productos es el perfil de sulfatación de tirosinas; Y1680 es la más importante ya que es esencial para la interacción con el VWF. A diferencia del pdFVIII, hasta 8% de las tirosinas de los rFVIII no está sulfatada, lo cual ocasiona que una proporción del FVIII no forme complejo con el VWF y, con ello, es blanco inmunológico.31 Además de las modificaciones postraduccionales, el contenido de sustancias distintas al FVIII en los derivados plasmáticos explicaría la diferente inmunogenicidad entre los productos.
En oposición de los productos recombinantes, los plasmáticos contienen VWF, fibronectina, inmunoglobulinas, fibrinógeno, albúmina, galectina 3, TGF-b, marcadores de activación de neutrófilos, plaquetas, moléculas del complemento, péptido de neutrófilo humano (HNP1-3), mieloperoxidasa, factor plaquetario 4, trombospondina 1 y C3a, entre otros componentes32,33, los cuales contribuirían a disminuir la inmunogenicidad del pdFVIII a partir del fenómeno de «competencia antigénica», según el cual, la exposición simultánea a múltiples antígenos puede reducir la respuesta inmune protectora colectiva por sobrecarga inmune.34
En cuanto a factores de riesgo asociados al producto se incluye la presencia de agregados de FVIII. Hay dos tipos de agregados, los no covalentes (también llamados nativos) y los no nativos en los cuales el monómero de FVIII está parcialmente no plegado.35 Los productos recombinantes contienen aproximadamente 20% de agregados de FVIII.36 La inmunogenicidad de los agregados de rFVIII es superior a la forma nativa del mismo producto; sin embargo, la inmunogenicidad de mezclas del FVIII nativo con 5% o 20% de agregados es menor, comparado con FVIII nativo.36,37 De manera opuesta a los agregados nativos de rFVIII, los no nativos son menos inmunogénicos que la forma nativa. De igual manera, las mezclas al 5% o al 20% de agregados son menos inmunogénicas que el FVIII nativo.37 Esto sugiere que la presencia de agregados nativos y no nativos al 5% o 20% en los concentrados plasmáticos disminuye la inmunogenicidad del FVIII por mecanismos desconocidos.
La diferencia entre la actividad real de FVIII y la informada por el fabricante puede ocasionar sobre o subdosificación del FVIII. En el primer caso, tiene lugar la exposición excesiva del factor ante el sistema inmune y, en el segundo, la falta de eficacia terapéutica y hemorragia.33 La importancia de esto radica en que, por sí sola, la sobredosificación de FVIII es un factor de riesgo para desarrollar inhibidores. Sabemos que, comparando una dosis media de 35 UI/kg por 5 días, el riesgo de generar inhibidores es 2.4 veces mayor que al usar una dosis promedio de 35-50 UI/kg y 2.3 veces mayor si la dosis es >50 UI/kg.
En cuanto al tipo de tratamiento, sabemos que en los pacientes que inician profilaxis, durante los primeros 75 días de exposición al FVIII, el riesgo de desarrollar inhibidores es 32% más bajo, en comparación con los tratados a demanda. Además, entre pacientes en profilaxis, el riesgo de inhibidores es 31% más bajo cuando el tratamiento inicia en los primeros 15 días de exposición al FVIII. No existe una relación entre la dosis, la frecuencia de la profilaxis y el desarrollo de inhibidores.27,38 En conclusión, el tratamiento con dosis altas de FVIII incrementa el riesgo de inhibidores, mientras que el tratamiento profiláctico lo disminuye.38
Respuesta inmune anti-FVIII
La respuesta inmune contra el FVIII es de tipo T dependiente. Una respuesta inmune clásica dependiente de linfocitos T empieza cuando una célula presentadora de antígeno (CPA) inmadura encuentra señales de daño o peligro, lo que la hace madurar, es decir, la expresión de complejos MHCII-péptido aumenta, lo mismo ocurre con las moléculas coestimuladoras y citocinas.39
Posteriormente, las CPA maduras presentan complejos MHC-péptido a linfocitos T; los linfocitos que sean específicos para el péptido presentado se activarán y diferenciarán hacia los distintos perfiles de estas células dependiendo del antígeno y del microambiente de citocinas. La respuesta inmune se lleva a cabo tanto en sitios de lesión vascular como en órganos linfoides secundarios. El bazo tiene un papel fundamental en el desarrollo de la inmunidad especialmente contra antígenos de la sangre, los cuales llegan al bazo por la arteria esplénica, la cual se bifurca hacia la pulpa roja e interactúa con los macrófagos de esta zona o hacia la zona marginal (ZM) donde residen células B de zona marginal (BZM), macrófagos metalofílicos y macrófagos de la zona marginal (MZM) (Figura 1).40
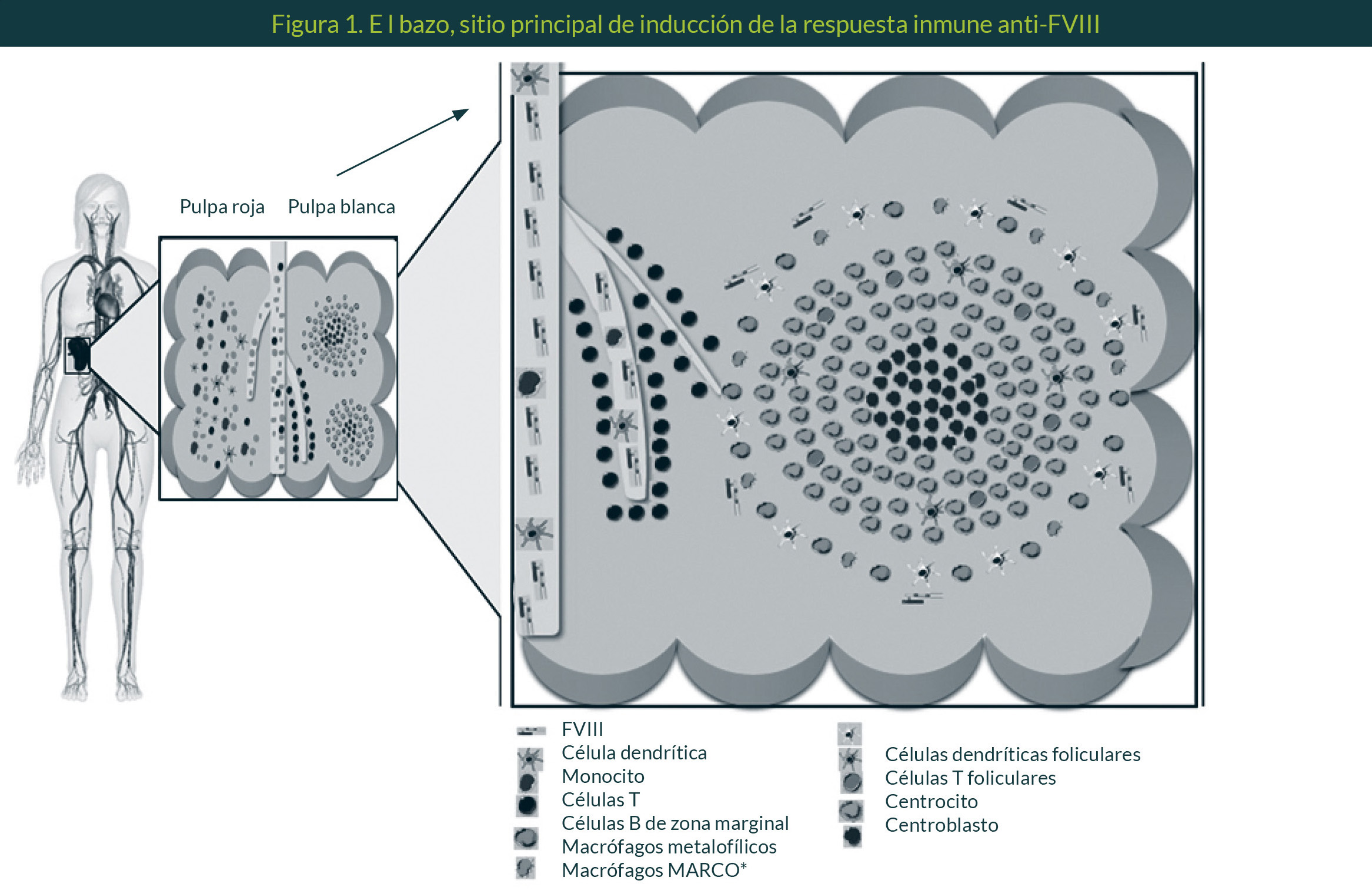
Para conocer el lugar y las células participantes en la inducción de la respuesta anti-FVIII, se inyectó FVIII en un modelo múrido de HA y se investigó su biodistribución. El factor se acumuló en el bazo a la par que su concentración plasmática disminuye. El factor se localiza en la ZM, preferentemente, en los macrófagos metalofílicos (MOMA1+), macrófagos MARCO+ y células dendríticas (DC) CD11c+40, pero también en BZM.41
Debido a que las BZM transportan antígenos opsonizados por complemento a las células dendríticas foliculares (fDC), se estudió la influencia del complemento en la endocitosis del FVIII. La evidencia muestra que la endocitosis del FVIII puede estar mediada por receptores antígeno específicos (como el BCR) o receptores «antígeno no específico» (como el receptor de manosas (CD206 /CD91), y que es facilitada por su unión a la molécula C3b del complemento.42
Dado que el FVIII circula unido al VWF, se investigó la influencia de este último en la endocitosis y presentación de péptidos del FVIII en el contexto de las moléculas de histocompatibilidad de clase II. El VWF reduce la interacción del FVIII con las células dendríticas inmaduras (iDC) y su subsecuente endocitosis, además, en presencia del VWF, el repertorio de péptidos del FVIII presentados se altera, en comparación con los péptidos encontrados en las iDC estimuladas únicamente con FVIII.43
Por otro lado, ahora se conoce el microambiente de citocinas generado en torno al FVIII que contribuye a la polarización de la respuesta celular T hacia un perfil Th1 o Th2. Se ha demostrado que neutrófilos de pacientes con inhibidor producen menos TNFa e IL-4, pero más IL-10 que los de pacientes sin inhibidor. Además, los monocitos de pacientes con inhibidor producen menos TNFa e IL-5 que los de pacientes sin inhibidor44,45; que los linfocitos T CD4+ de pacientes con inhibidores producen menos TNFa, IL-4, IL-5 que los de pacientes sin inhibidor; y que los linfocitos T CD8+ de pacientes con inhibidor producen menos IFNg, IL-4, IL-5 y TNFa que los de pacientes sin inhibidor. Ambas poblaciones de linfocitos de pacientes con inhibidor producen más IL-1043,44 y que el perfil de citocinas producto de los tres tipos celulares es igual para pdFVIII y rFVIII.44,45
Estos resultados sugieren predominio de un perfil antinflamatorio regulador en hemofílicos con inhibidor y un patrón mixto hacia lo proinflamatorio en pacientes sin inhibidor, modulado por IL-4 e IL-545: el perfil TH2 aparece en pacientes con inhibidores y el TH1 en hemofílicos sin inhibidores. Además de estos perfiles, la subclase de IgG es diferente entre pacientes con y sin inhibidor: los anticuerpos IgG4 predominan en pacientes con inhibidor y las IgG1 en enfermos sin inhibidor45. Asimismo, en pacientes con inhibidor aumenta IL-10 y disminuye IL-4, lo cual se explica por el efecto de IL-10 sobre las población de linfocitos TH2 en los cuales evita la sobreproducción de IL-4,IL-5 e IL-13.46
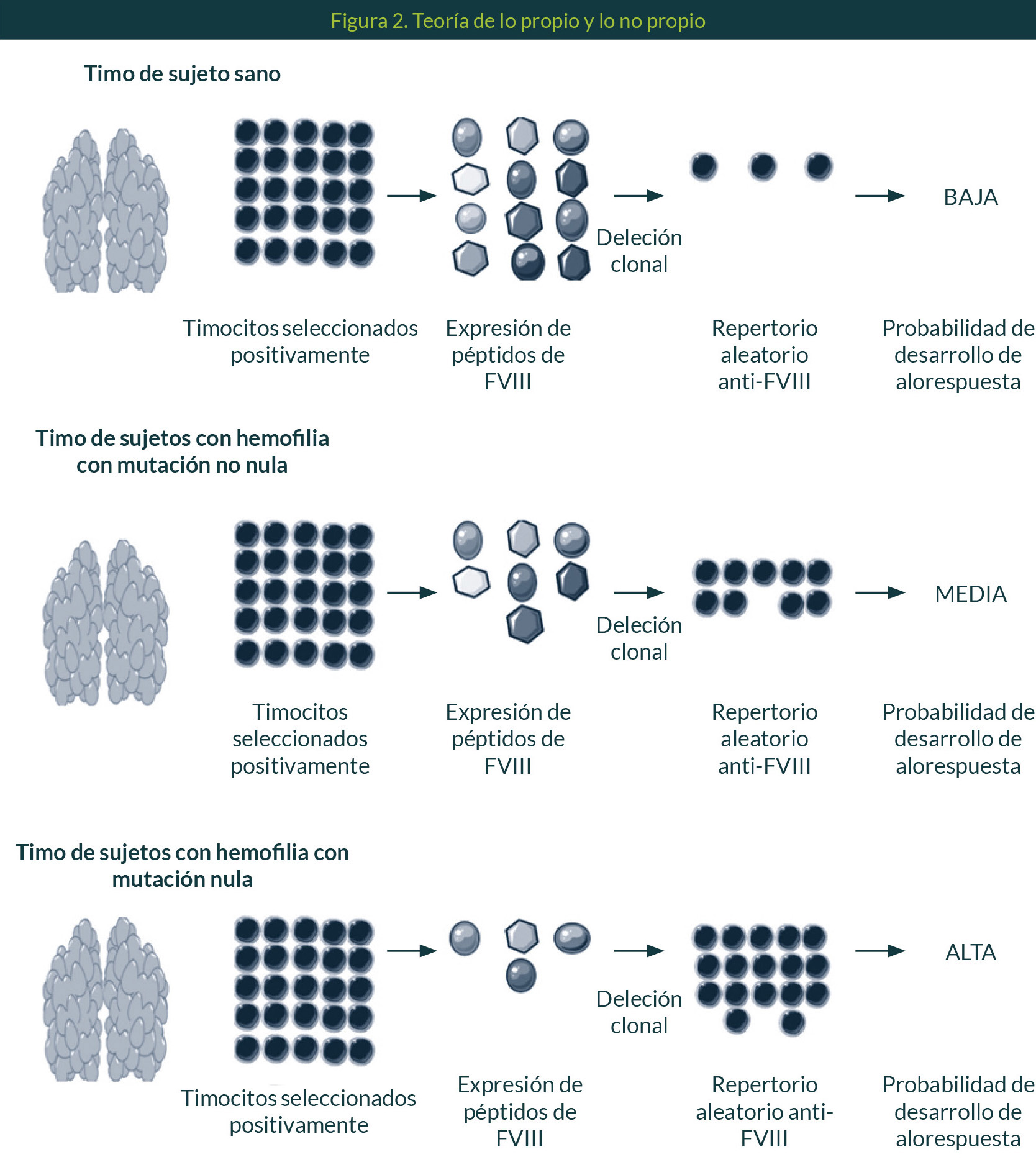
De acuerdo con la «teoría del daño», la respuesta inmune se activa debido a la emisión de señales de daño. En contraste con la «teoría de lo propio y lo no propio», la respuesta inmune es activada en contra de entidades ajenas mientras que no se desencadena una respuesta inmune en contra de los constituyentes propios (Figura 2).47
En oposición a la teoría de lo propio y no propio, la teoría del daño propone que los constituyentes propios pueden activar la respuesta inmune si ellos son peligrosos (Figura 3). Esto puede aparecer por estrés celular o por moléculas asociadas a daño generadas en algunos injertos. Entonces, algunos constituyentes no propios pueden ser tolerados si no son peligrosos (fetos, bacterias comensales).48 Ejemplos de señales de daño pueden ser citocinas como CD40L, TNFa, IL1b, IFNa; componentes intracelulares como ATP, UTP; proteínas de choque térmico, componentes del inflamosoma o actina F, entre muchas otras.47
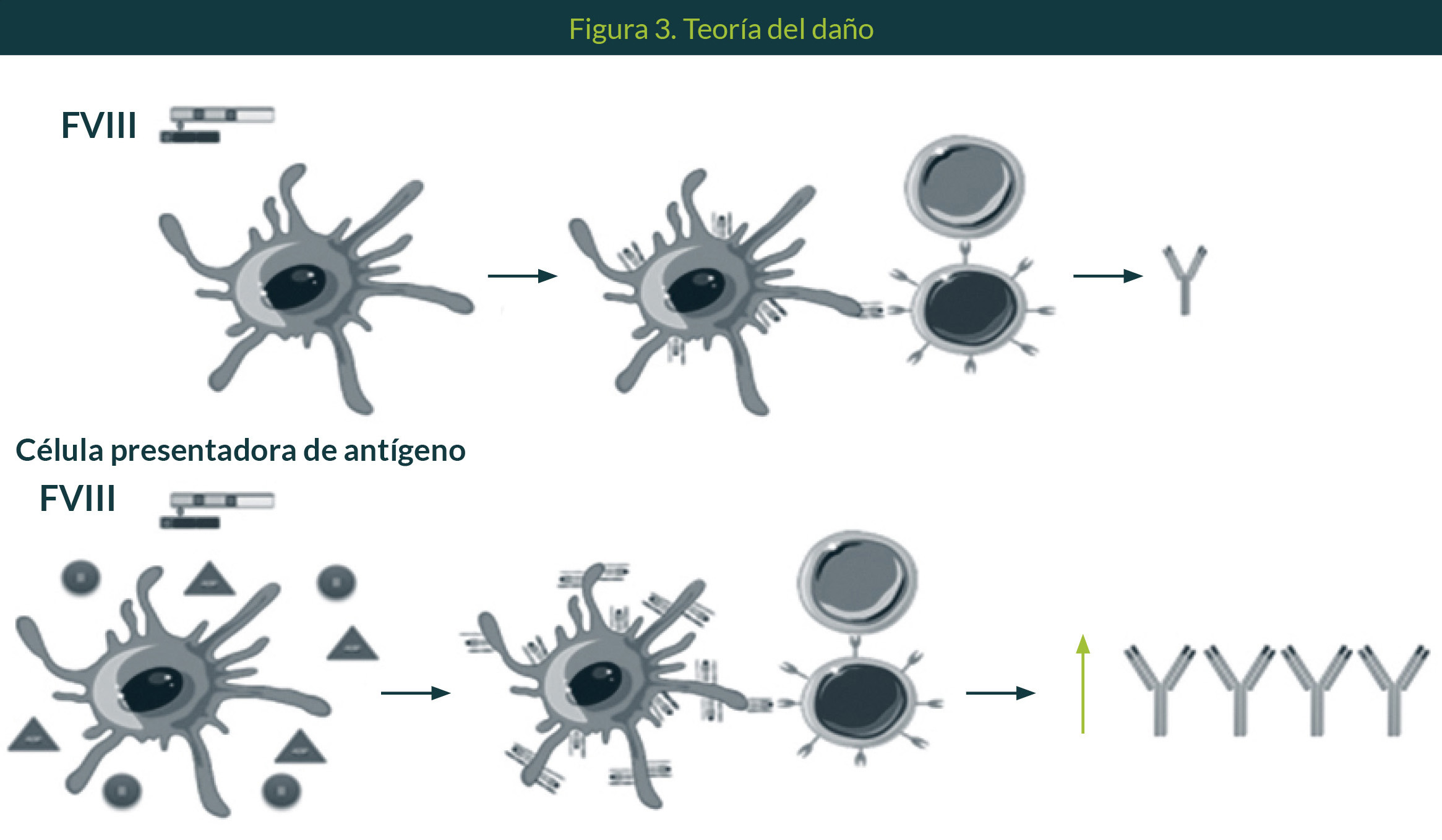
El postulado principal de la teoría del daño es que no hay activación de la respuesta inmune sin presencia de daño48; y el de la teoría de lo propio y lo no propio es que la eliminación de clonas autoreactivas protege al cuerpo de lo que Paul Ehrlich llamó horror autotoxicus.49 Pero entonces, ¿cuál teoría explica la respuesta inmune contra el FVIII en los pacientes con HA grave?
En los siguientes párrafos se demuestra con evidencia científica que ambas teorías son correctas y no necesariamente excluyentes en el contexto del desarrollo de respuesta inmune anti-FVIII en pacientes con HA.
Por ejemplo, el estudio SIPPET concluyó que el rFVIII es más inmunogénico que pdFVIII29; sin embargo, esto no concuerda con la teoría del daño ya que la incubación de DC inmaduras en presencia de rFVIII no induce la maduración de éstas. En contraste, la incubación de DC inmaduras en presencia de pdFVIII sí causa la maduración de las DC, medido a través de la expresión de moléculas de coestimulación como CD83 y CD86 y de TNFa, IL6, IL12, IL10 e IL5.50,51 La maduración de las iDC por pdFVIII puede deberse a que componentes como HNP 1-3, mieloperoxidasa, factor plaquetario 4, trombospondina y C3a del complemento32 son reconocidos como señales de daño. Lo anterior pone en contraste la teoría del daño con la realidad porque, de ser así, el pdFVIII debería ser más inmunogénico que el rFVIII.
Por otro lado, el postulado principal de la teoría de lo propio y lo no propio es que la eliminación de clonas autorreactivas protege al cuerpo del horror autotoxicus al cual hoy podríamos llamarle enfermedad autoinmune. Sin embargo, en el contexto de los pacientes con HA tratados con FVIII, la respuesta contra esta proteína se lleva a cabo contra un antígeno no propio. Si se lograra que el sistema inmune reconociera al FVIII como propio y disminuyeran las señales de daño podría evitarse la producción de inhibidores. Por lo tanto, surge la pregunta: ¿cómo lograr que el sistema inmune del paciente con HA vea al FVIII exógeno como propio?
Al respecto, la evidencia muestra que la inoculación intratímica de FVIII, a ratones deficientes de este factor, provoca niveles bajos o indetectables de anticuerpos anti-FVIII, incluso después de refuerzos continuos con FVIII en adyuvante de Freund. Debido a que las células T CD4+ aisladas de estos ratones no proliferan ni producen citocinas inflamatorias (a excepción de IL4) en respuesta a la estimulación con FVIII y coincubación con APC, se sugiere que la respuesta TH1 anti-FVIII se suprime. Además, el aumento de las células T reguladoras CD4+ CD25+ FoxP3+ en estos ratones permite hipotetizar que el mecanismo de supresión de la respuesta inmune anti-FVIII es mediado por estas células.52 En adición a las células T reguladoras, existe la posibilidad de que la supresión de la respuesta inmune anti-FVIII esté mediada por las DC tímicas, ya que, entre las funciones de estas células se encuentra la eliminación de clonas aloreactivas. Sin embargo, esta posibilidad no fue evaluada en la investigación.52
En comparación con los pacientes tratados a demanda, los pacientes en profilaxis tienen 32% menos riesgo de desarrollar inhibidor.38 En el contexto de la teoría del daño, esto podría traducirse como que, en ausencia de señales de daño (hemorragia o inflamación), el riesgo de desarrollar inhibidor disminuye, aunque señala que, aún en ausencia de señales de daño, el paciente desarrolla anticuerpos. La pregunta es: ¿el FVIII es intrínsecamente inmunogénico o la inmunogenicidad depende de su actividad procoagulante? Se sabe que ratones deficientes de FVIII producen títulos de anticuerpos 100 veces más altos que los tratados con ovoalbúmina y que la dosis de FVIII que induce la respuesta inmune es 50 veces menor vs. otros antígenos.53 Esto sugiere que el FVIII es una proteína intrínsecamente inmunogénica; no obstante, su inmunogenicidad se acentuó cuando, a partir de resultados obtenidos de ratones KO y del uso de mutantes de FVIII que modifican la actividad del factor, se demostró que la respuesta inmune anti-FVIII depende de la estructura de la proteína y no de su actividad.53 Esto explicaría porque los pacientes con HA con deleciones en múltiples exones del F8 y/o con mutaciones nulas de este gen tienen más riesgo de desarrollar inhibidor que aquellos con mutaciones con sentido equivocado y/o mutaciones no nulas.54-56 Además, sustenta la aplicación de la teoría de lo propio y no propio en el entendimiento de la respuesta inmune anti-FVIII.
Retomando la teoría del daño, se tiene evidencia de que, en el contexto de un estado inflamatorio, la generación de la respuesta inmune anti-FVIII aumenta en comparación con un estado basal. Esto fue demostrado al simular una inflamación viral en un modelo múrido de HA al administrar PIC (adyuvante que induce inflamación parecida a viral). Los ratones con HA expuestos a FVIII en presencia de PIC mostraron títulos más altos de anticuerpos anti-FVIII que los receptores que sólo recibieron FVIII con solución salina. Ello sugiere que el estado inflamatorio subyacente podría afectar en la magnitud de la respuesta de anticuerpos.41
Estudios anteriores sugerían que el FVIII es intrínsecamente más inmunogénico que los antígenos modelo, que su inmunogenicidad disminuía en presencia de VWF y que aumentaba en presencia de inflamación parecida a viral. No obstante, la inflamación parecida a la de la respuesta a un virus no es una situación hemostáticamente común; por ello, se plantea si el estado procoagulante desencadenado por la administración de FVIII actúa como adyuvante natural de la respuesta anti-FVIII.
Entre los hallazgos encontrados destaca que la anticoagulación con warfarina, administrada a ratones deficientes de FVIII tratados con FVIII, conlleva una respuesta humoral y celular significativamente menor que la de ratones no anticoagulados, evaluadas mediante proliferación de linfocitos T y medición de los títulos de anticuerpos.39 Lo mismo sucedió al inhibir selectivamente la trombina con hirudina, con lo cual se evidenció que la trombina tiene propiedades adyuvantes.39 Sin embargo, la disminución en la respuesta humoral es mayor con warfarina vs. hirudina, lo que sugiere que otros factores hemostáticos diferentes a trombina aumentan la inmunogenicidad del FVIII.39
No obstante, la anticoagulación del paciente con HA grave es irreal y lo es aún más la inhibición directa de la trombina; entonces, se han buscado elementos farmacológicos para disminuir la producción de patrones moleculares asociados a patógenos (PAMP, pathogen-associated molecular patterns) o patrones moleculares asociados a daño (DAMP, damage associated molecular patterns) y así evitar el ambiente inflamatorio y el consecuente aumento de la inmunogenicidad del FVIII. Una posibilidad para evitar las señales proinflamatorias podrían ser los glucocorticoides porque suprimen las señales proinflamatorias con consecuencias inmunológicas. La dexametasona se ha evaluado en ratones deficientes de FVIII. Al coadministrarse con FVIII disminuye la incidencia y el título de inhibidores, en comparación con ratones control.57 Se analizó su papel en la tolerancia hacia el FVIII y se encontró que al exponer a ratones previamente tratados con dexametasona y FVIII, a FVIII + LPS, 7% desarrolló inhibidor vs. 52% de los tratados sólo con FVIII. Además, el título de anticuerpos también disminuyó. El efecto permanece por al menos 18 semanas y, ya que el tratamiento con FVIII y dexametasona induce aumento de la población de células T reguladoras CD4+ CD8- CD25+ FoxP3+, se propone a estas células como responsables de la tolerancia al FVIII.57
Con base en estos resultados, podría sugerirse el uso de dexametasona concomitantemente al tratamiento a demanda con FVIII para disminuir la respuesta anti-FVIII al inhibir la presentación antigénica y promover la expresión de citocinas antinflamatorias.58 Esto se aplicaría únicamente bajo un contexto inflamatorio, tal y como ocurre en la hemorragia.
Otras estrategias propuestas para disminuir la inmunogenicidad del FVIII son administrar FVIII de vida media más larga pegilado o fusionado con fracciones cristalizables de IgG1.59 Desde el punto de vista de la terapia celular, se podrían administrar células CART con fenotipo de células T reguladoras específicas de FVIII capaces de suprimir la respuesta anti-FVIII, o bien60, administrar DC preincubadas con FVIII, incapaces de expresar moléculas de coestimulación para que, de esta manera, bajo los preceptos de la primera ley de los linfocitos, las células capaces de reconocer péptidos de FVIII en el contexto del MHC clase II mueran en ausencia de coestimulación.48
En conclusión, la respuesta inmune contra el FVIII es una respuesta de tipo T dependiente con un perfil TH2 en hemofílicos con inhibidores y perfil Th1 en los que no lo desarrollan. Sobre la base de las teorías del daño y de lo propio y no propio, la inmunogenicidad del FVIII depende principalmente de la mutación del F8, del contexto o microambiente en el que se administra el FVIII (inflamatorio y no inflamatorio) y del origen del factor (plasmático o recombinante). Dicho de otro modo, en un contexto inflamatorio, la inmunogenicidad del factor aumentará; por el contrario, en la homeostasia, la inmunogenicidad será intrínseca a la estructura y origen del FVIII (los productos recombinantes son más inmunogénicos), y disminuirá por influencia del VWF. Visto desde otra perspectiva, puede decirse que los factores de riesgo de inhibidores asociados al paciente, como las mutaciones del F8 y el haplotipo del FVIII, podrían relacionarse con la «teoría de lo propio y lo no propio»; los factores de riesgo asociados al tratamiento, como la dosis y el origen de FVIII, se relacionarían con la «teoría del daño».
Ninguno que declarar.
| 1. | Ragni MV, Berntorp E, Carcao M, Escuriola Ettingshausen C, Nedzinskas A, Ozelo MC, et al. Inhibitors to clotting factor. Haemophila. 2020;95-105. |
| 2. | Bhopale GM, Nanda RK. Blood coagulation factor VIII: An overview. J Biosci. 2003;28(6):783-9. |
| 3. | Garcia-Chávez J, Majluf-Cruz A. Hemofilia. Gac Med Mex. 2013;149(3):308–21. |
| 4. | Lenting PJ, Christophe OD, Guéguen P. The disappearing act of factor VIII. Haemophilia. 2010;16(102):6-15. |
| 5. | Pipe SW. Functional roles of the factor VIII B domain. Haemophilia. 2009;15(6):1187-96. |
| 6. | Shahani T, Covens K, Lavend'homme R, Jazouli N, Sokal E, Peerlinck K, et al. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J Thromb Haemost. 2014;12(1):36-42. |
| 7. | Orlova NA, Kovnir SV, Vorobiev II, Gabibov AG, Vorobiev AI. Blood Clotting Factor VIII: From Evolution to Therapy. Acta Naturae. 2013;5(2):19-39. |
| 8. | Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361(9371):1801-9. |
| 9. | Keeling D, Tait C, Makris M. Guideline on the selection and use of therapeutic products to treat haemophilia and other hereditary bleeding disorders. A United Kingdom Haemophilia Center Doctors' Organisation (UKHCDO) guideline approved by the British Committee for Standards in Haematology. Haemophilia. 2008;14(4):671-84. |
| 10. | Aznar JA, Lucía F, Abad-Franch L, Jiménez-Yuste V, Pérez R, Batlle J, et al. Haemophilia in Spain. Haemophilia. 2009;15(3):665-75. |
| 11. | Coppola A, Franchini M, Tagliaferri A. Prophylaxis in people with haemophilia. Thromb Haemost. 2009;101(4):674-81. |
| 12. | Nilsson IM, Berntorp E, Löfqvist T, Pettersson H. Twenty-five years' experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232(1):25-32. |
| 13. | Berntorp E, Boulyjenkov V, Brettler D, Chandy M, Jones P, Lee C, et al. Modern treatment of haemophilia. Bull World Health Organ. 1995;73(5):691-701. |
| 14. | Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M, Pipe SW, et al. Guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26 Suppl 6:1-158. |
| 15. | Geraghty S, Dunkley T, Harrington C, Lindvall K, Maahs J, Sek J. Practice patterns in haemophilia A therapy- global progress towards optimal care. Haemophilia. 2006;12(1):75-81. |
| 16. | Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535-44. |
| 17. | Jiménez-Yuste V, Alvarez MT, Martín-Salces M, Quintana M, Rodriguez-Merchan C, Lopez-Cabarcos C, et al. Prophylaxis in 10 patients with severe haemophilia A and inhibitor: Different approaches for different clinical situations. Haemophilia. 2009;15(1):203-9. |
| 18. | Ahlberg A. Haemophilia in Sweden. VII. Incidence, treatment and prophylaxis of arthropathy and other musculo-skeletal manifestations of haemophilia A and B. Acta Orthop Scand Suppl. 1965:(suppl 77):3-132. |
| 19. | Aznar JA, Moret A, Haya S, Cid AR, Cabrera N, Abad L, et al. Fisiopatología y diagnóstico de los inhibidores del FVIII. In: Yuste VJ, ed. Inhibidores en Hemofilia. Madrid, España: Momento Médico Iberoamericana; 2009:57–70. |
| 20. | Lewis KB, Hughes RJ, Epstein MS, Josephson NC, Kempton CL, Kessler CM, et al. Phenotypes of allo- and autoimmune antibody responses to FVIII characterized by surface plasmon resonance. PLoS One. 2013;8(5):e61120. |
| 21. | Whelan SF, Hofbauer CJ, Horling FM, Allacher P, Wolfsegger MJ, Oldenburg J, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. 2013;121(6):1039-48. |
| 22. | Wootla B, Dasgupta S, Dimitrov JD, Bayry J, Lévesque H, Borg JY, et al. Factor VIII hydrolysis mediated by anti-factor VIII autoantibodies in acquired hemophilia. J Immunol. 2008;180(11):7714-20. |
| 23. | Miller CH, Benson J, Ellingsen D, Driggers J, Payne A, Kelly FM, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18(3):375-82. |
| 24. | Schwarz J, Astermark J, Menius ED, Carrington M, Donfield SM, Gomperts ED, et al. F8 haplotype and inhibitor risk: Results from the Hemophilia Inhibitor Genetics Study (HIGS) combined cohort. Haemophilia. 2013;19(1):113-8. |
| 25. | Viel KR, Ameri A, Abshire TC, Iyer RV, Watts RG, Lutcher C, et al. Inhibitors of factor VIII in black patients with hemophilia. N Engl J Med. 2009;360(16):1618-27. |
| 26. | Franchini M, Coppola A, Mengoli C, Rivolta GF, Riccardi F, Minno GD, et al. Blood group O protects against inhibitor development in severe Hemophilia A patients. Semin Thromb Hemost. 2017;43(1):69-74. |
| 27. | Gouw SC, van der Bom JG, Marijke van den Berg H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: The CANAL cohort study. Blood. 2007;109(11):4648-54. |
| 28. | Cannavò A, Valsecchi C, Garagiola I, Palla R, Mannucci PM, Rosendaal FR, et al. Nonneutralizing antibodies against factor VIII and risk of inhibitor development in severe hemophilia A. Blood. 2017;129(10):1245-50. |
| 29. | Peyvandi F, Mannucci PM, Garagiola I, El-Beshlawy A, Elalfy M, Ramanan V, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054-64. |
| 30. | Canis K, Anzengruber J, Feichtinger M, Benamara K, Scheiflinger F, Savoy A, et al. In-depth comparison of N-glycosylation of human plasma- derived factor VIII and different recombinant products: From structure to clinical implications. J Thromb Haemostasis. 2018;16:1592-603. |
| 31. | Kannicht C, Ramström M, Kohla G, Tiemeyer M, Casademunt E, Walter O, et al. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res. 2013;131(1):78-88. |
| 32. | Brodde MF, Kehrel BE. Markers of blood cell activation and complement activation in factor VIII and von Willebrand factor concentrates. Transfus Med Hemother. 2010;37(4):175-84. |
| 33. | Pahl S, Pavlova A, Driesen J, Müller J, Pötzsch B, Oldenburg J. In vitro characterization of recombinant factor VIII concentrates reveals significant differences in protein content, activity and thrombin activation profile. Haemophilia. 2013;19(3):392-8. |
| 34. | Lai J, Hough C, Tarrant J, Lillicrap D. Biological considerations of plasma-derived and recombinant factor VIII immunogenicity. Blood. 2017;129(24):3147-54. |
| 35. | Pisal DS, Kosloski MP, Middaugh CR, Bankert RB, Balu-Iyer SV. Native-like aggregates of Factor VIII (FVIII) are immunogenic von Willebrand Factor deficient and hemophilia A mice. J Pharm Sci. 2012;101(6):2055-65. |
| 36. | Healey JF, Parker ET, Lollar P. Identification of aggregates in therapeutic formulations of recombinant full-length factor VIII products by sedimentation velocity analytical ultracentrifugation. J Thromb Haemost. 2018;16(2):303-15. |
| 37. | Purohit VS, Middaugh CR, Balasubramanian SV. Influence of aggregation on immunogenicity of recombinant human Factor VIII in hemophilia A mice. J Pharm Sci. 2006;95(2):358-71. |
| 38. | Gouw SC, van den Berg HM, Fischer K, Auerswald G, Carcao M, Chalmers E, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: The RODIN study. Blood. 2013;121(20):4046-55. |
| 39. | Skupsky J, Zhang AH, Su Y, Scott DW. A role for thrombin in the initiation of the immune response to therapeutic factor VIII. Blood. 2009;114(21):4741-8. |
| 40. | Navarrete A, Dasgupta S, Delignat S, Caligiuri G, Christophe OD, Bayry J, et al. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J Thromb Haemost. 2009;7(11):1816-23. |
| 41. | Zerra PE, Cox C, Baldwin WH, Patel SR, Arthur CM, Lollar P, et al. Marginal zone B cells are critical to factor VIII inhibitor formation in mice with hemophilia A. Blood. 2017;130(23):2559-68. |
| 42. | Rayes J, Ing M, Delignat S, Peyron I, Gilardin L, Vogel CW, et al. Complement C3 is a novel modulator of the anti-factor VIII immune response. Haematologica. 2018;103(2):351-60. |
| 43. | Sorvillo N, Hartholt RB, Bloem E, Sedek M, ten Brinke A, van der Zwaan C, et al. von Willebrand factor binds to the surface of dendritic cells and modulates peptide presentation of factor VIII. Haematologica. 2016;101(3):309-18. |
| 44. | Silveira AC, Santana MA, Ribeiro IG, Chaves DG, Martins-Filho OA. The IL-10 polarized cytokine pattern in innate and adaptive immunity cells contribute to the development of FVIII inhibitors. BMC Hematol. 2015;15(1):1. |
| 45. | Chaves DG, Velloso-Rodrigues C, Oliveira CA, Teixeira-Carvalho A, Santoro MM, Martins-Filho OA. A shift towards a T cell cytokine deficiency along with an anti-inflammatory/regulatory microenvironment may enable the synthesis of anti-FVIII inhibitors in haemophilia A patients. Clin Exp Immunol. 2010;162(3):425-37. |
| 46. | Couper KN, Blount DG, Riley EM. IL-10: The master regulator of immunity to infection. J Immunol. 2008;180(9):5771-7. |
| 47. | Pradeu T, Cooper EL. The danger theory: 20 years later. Front Immunol. 2012;3:287. |
| 48. | Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991-1045. |
| 49. | Tauber AI, Podolsky SH. Frank Macfarlane Burnet and the immune self. J Hist Biol. 1994;27(3):531-73. |
| 50. | Miller L, Weissmüller S, Ringler E, Crauwels P, van Zandbergen G, Seitz R, et al. Danger signal-dependent activation of human dendritic cells by plasma-derived factor VIII products. Thromb Haemost. 2015;114(2):268-76. |
| 51. | Pfistershammer K, Stöckl J, Siekmann J, Turecek PL, Schwarz HP, Reipert BM. Recombinant factor VIII and factor VIII-von Willebrand factor complex do not present danger signals for human dendritic cells. Thromb Haemost. 2006;96(3):309-16. |
| 52. | Madoiwa S, Yamauchi T, Kobayashi E, Hakamata Y, Dokai M, Makino N, et al. Induction of factor VIII-specific unresponsiveness by intrathymic factor VIII injection in murine hemophilia A. J Thromb Haemost. 2009;7(5):811-24. |
| 53. | Meeks SL, Cox CL, Healey JF, Parker ET, Doshi BS, Gangadharan B, et al. A major determinant of the immunogenicity of factor VIII in a murine model is independent of its procoagulant function. Blood. 2012;120(12):2512-20. |
| 54. | Gouw SC, van den Berg HM, Oldenburg J, Astermark J, de Groot PG, Margaglione M, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: Systematic review and meta-analysis. Blood. 2012;119(12):2922-34. |
| 55. | Carcao MD, van den Berg HM, Ljung R, Mancuso ME. Correlation between phenotype and genotype in a large unselected cohort of children with severe hemophilia A. Blood. 2013;121(19):3946-52. |
| 56. | Spena S, Garagiola I, Cannavò A, Mortarino M, Mannucci PM, Rosendaal FR, et al. Prediction of factor VIII inhibitor development in the SIPPET cohort by mutational analysis and factor VIII antigen measurement. J Thromb Haemost. 2018;16(4):778-90. |
| 57. | Georgescu MT, Moorehead PC, van Velzen AS, Nesbitt K, Reipert BM, Steinitz KN, et al. Dexamethasone promotes durable factor VIII-specific tolerance in hemophilia A mice via thymic mechanisms. Haematologica. 2018;103(8):1403-13. |
| 58. | Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. 2017;17(4):233–47. |
| 59. | Graf L. Extended half-Life factor VIII and factor IX preparations. Transfus Med Hemother. 2018;45(2):86-91. |
| 60. | Parvathaneni K, Abdeladhim M, Pratt KP, Scott DW. Hemophilia A inhibitor treatment: The promise of engineered T-cell therapy. Transl Res. 2017;187:44-52. |
All Rights Reserved® 2019
Latin American Journal of Clinical Sciences and Medical Technology,Publicación contínua • Editor responsable: Gilberto Castañeda Hernández. • Reserva de Derechos al Uso Exclusivo: 04-2019-062013242000-203; ISSN: 2683-2291; ambos otorgados por el Instituto Nacional del Derecho de Autor. • Responsable de la última actualización de este número, Web Master Hunahpú Velázquez Martínez,
Calle Profesor Miguel Serrano #8, Col. Del Valle, Alcaldía Benito Juárez, CP 03100, Ciudad de México, México. Número telefónico: 55 5405 1396 • Fecha de última modificación, 28 de agosto de 2024.
All Rights Reserved® 2019
Publicación contínua • Editor responsable: Gilberto Castañeda Hernández. • Reserva de Derechos al Uso Exclusivo: 04-2019-062013242000-203; ISSN: 2683-2291; ambos otorgados por el Instituto Nacional del Derecho de Autor. • Responsable de la última actualización de este número, Web Master Hunahpú Velázquez Martínez,
Calle Profesor Miguel Serrano #8, Col. Del Valle, Alcaldía Benito Juárez, CP 03100, Ciudad de México, México. Número telefónico: 55 5405 1396 • Fecha de última modificación, 28 de agosto de 2024.