| ** | Latin American Journal of Clinical Sciences and Medical Technology is an open access magazine. To read all published articles and materials you just need to register Registration is free of charge. Register now If you already have registered please Log In | ** |




 0009-0007-1885-5348)a; José Eduardo Juárez-Hernández (0000-0002-1882-9816)a; Lourdes González-Flores a; Leticia Cruz-Antonio (0000-0001-8812-9711)b.
0009-0007-1885-5348)a; José Eduardo Juárez-Hernández (0000-0002-1882-9816)a; Lourdes González-Flores a; Leticia Cruz-Antonio (0000-0001-8812-9711)b.aDepartamento de Farmacología, Centro de Investigación y de Estudios Avanzados, Instituto Politécnico Nacional, Ciudad de México, México; bFacultad de Estudios Superiores, Zaragoza, UNAM, Ciudad de México, México.
Autor para correspondencia: , . Números telefónicos: ; e-mail: letycruza@yahoo.com.mx

Lat Am J Clin Sci Med Technol. 2024 Jul;6:138-147.
Recibido: 09 de abril, 2024
Aceptado: 22 de junio, 2024
Publicado: 23 de julio, 2024
Vistas: 2003
Descargas: 10

Diversos organismos de normalización de diferente naturaleza indican con claridad cómo obtener la evidencia experimental documentada que certifica la validez de los métodos analíticos para cuantificar fármacos en matriz biológica para estudios de bioequivalencia. La vasta información respecto a este tema resalta la relevancia de uniformar los procedimientos para lograr el objetivo deseado, lo cual puede ser aplicado por cualquier laboratorio que lo requiera. Sin embargo, para evitar que por legislación local existan discrepancias en cuanto a la interpretación y/o ejecución de las diversas formas de evaluación de desempeño analítico por parte de la autoridad respecto a la información internacional proporcionada, se emiten regulaciones locales con carácter de obligatorias que resaltan situaciones trascendentales con criterios discriminatorios. Este trabajo presenta las características mínimas de desempeño analítico y los criterios de aceptación que se requieren, acorde con la normatividad mexicana, para demostrar que un método analítico es válido para cuantificar fármacos en matriz biológica por CLAR (cromatografía líquida de alta resolución) para un estudio de investigación en una institución educativa a nivel posgrado. Se resalta que los parámetros analíticos a cumplir bajo esta normatividad consideran los aspectos específicos para la técnica y muestra biológica. Para ello, se emplea (a modo de ejemplo) un método desarrollado y validado por nuestro grupo de investigación para cuantificar lamotrigina en plasma humano con fines de monitoreo. Se concluye que las características de desempeño de la NOM-177-SSA-2013 aplicadas a la validación del método desarrollado evidencian la validez y aplicación del método para el objetivo deseado.
Standardization organizations of different natures indicate how to obtain documented experimental evidence that certifies the validity of analytical methods to quantify drugs in the biological matrix for bioequivalence studies. The vast amount of information on this subject highlights its relevance in uniforming the procedures to achieve the desired objective, which can be applied by any laboratory that requires it. However, to avoid discrepancies in the interpretation and/or execution of the various forms of analytical performance evaluation by the authority concerning the international information provided, local regulations are issued as mandatory, highlighting critical situations with discriminatory criteria. This paper presents the minimum analytical performance characteristics and the acceptance criteria required (according to Mexican regulations) to demonstrate that an analytical method is valid to quantify drugs in a biological matrix by HPLC (high-performance liquid chromatography) for a research study at a graduate-level educational institution. It is emphasized that the analytical parameters to be met under this standard consider the specific aspects of the technique and biological sample. For this purpose, a method developed and validated by our research group is used as an example for studying the biological sample. For this purpose, a method developed and validated by our research group to quantify lamotrigine in human plasma for monitoring purposes is used as an example. The performance characteristics of the NOM-177-SSA-2013 applied to the validation of the developed method demonstrate the validity and application of the method for the desired objective.
A medida que creció el interés por lograr uniformidad en el tipo y ejecución de parámetros analíticos para la validación de métodos donde las muestras a analizar son matrices biológicas, la literatura de diferentes organismos regulatorios o no al respecto ha sido variada y extensa.1-3
Aun cuando hoy en día se tiene un alto grado de uniformidad en la terminología, tipo y metodología de los parámetros aplicados para alcanzar la validación de métodos analíticos en matrices biológicas —que incluso alcanza la armonización del tema entre Estados Unidos de Norteamérica (FDA, Food and Drug Administration)4, la Unión Europea (EMA, European Medicines Agency)5 y Japón (ICH, International Council for Harmonisation)6— el por qué, cuándo, dónde y cómo aplicar las características de desempeño sigue dependiendo del alcance y la literatura consultada.
Demostrar que un método analítico es válido para estudios de biodisponibilidad, bioequivalencia y farmacocinética no sólo es una exigencia previa para contar con interpretación adecuada de los datos que promoverán decisiones clínicas en el tipo de estudios mencionados, también es requisito legal.
Para evitar que en un ámbito local exista discrepancia en cuanto a la interpretación y/o ejecución de los diversos parámetros analíticos —por parte de la autoridad respecto a la información internacional existente y llegue a ser un obstáculo técnico para alcanzar el objetivo deseado—, se emiten normas de cumplimiento obligatorio.
En México, existen varios organismos que proporcionan información útil para llevar a cabo la validación de un método analítico sin el alcance de la aplicación para métodos bioanalíticos, tales como las guías de validación del Colegio Nacional de Químicos Farmacéuticos Biólogos de México A.C.7, manuales de desarrollo analítico (UNAM)8 y la Farmacopea de los Estados Unidos Mexicanos (FEUM).9
Sin embargo, únicamente la Norma Oficial Mexicana 177-SSA1-201310 de cumplimiento obligatorio es el documento oficial vigente que abarca los requisitos mínimos y criterios de aceptación a los cuales deben apegarse los laboratorios, hospitales, centros de investigación, universidades e instituciones que llevan a cabo la validación de métodos analíticos para cuantificar fármacos en matriz biológica.
Este trabajo desglosa parte de la normatividad mexicana que orienta, simplifica y unifica criterios para demostrar que un método analítico es válido para cuantificar fármacos y/o sus metabolitos en una matriz biológica por CLAR (cromatografía líquida de alta resolución), aplicada a estudios de biodisponibilidad, bioequivalencia y/o farmacocinética sin contraponer su propósito a lo dispuesto por la normatividad internacional con un ejemplo específico: la validación de un método analítico para la cuantificación de lamotrigina en plasma humano desarrollado en un laboratorio de investigación farmacológica con fines de monitoreo clínico.
Al final se concluye que las características de desempeño de la NOM-177-SSA-2013, aplicadas a la validación del método desarrollado, evidencian la validez y aplicación de éste para los fines establecidos.
Reactivos
Para cuantificar lamotrigina en plasma humano se utilizó lamotrigina estándar secundario farmacéutico (Sigma-Aldrich) y como estándar interno metilparabeno (Supelco).
Para la preparación de la fase móvil se utilizaron acetonitrilo (Honeywell) y metanol (BDH) grado cromatográfico, así como fosfato monobásico de potasio (J.T. Baker).
El agua desionizada de alta pureza se obtuvo internamente mediante un equipo Milli-Q Regent Water System.
La fase móvil y disolventes para emplear en el sistema cromatográfico se filtraron a través de membrana de nylon (47 mm, tamaño de poro 0.45 μm, Sigma-Aldrich).
Preparación de soluciones
Fase móvil
Fue preparada con una mezcla de solución de fosfatos 0.03 M, pH 4.5, acetonitrilo y metanol en una proporción 70:20:10 v/v/v, respectivamente.
Se pesaron 0.005 g de lamotrigina estándar y se disolvieron con 20 mL de acetonitrilo en un matraz volumétrico de 50 mL. Una vez que se disolvió mediante un proceso de sonicación, se llevó a volumen con acetonitrilo.
De la solución stock A de lamotrigina se transfirieron 5 mL a un matraz volumétrico de 50 mL, y se llevó a volumen con acetonitrilo.
Se pesaron 0.01 g de metilparabeno y se disolvieron con 20 mL de metanol en un matraz volumétrico de 50 mL, para posteriormente llevar a aforo con el mismo disolvente.
Preparación de curva de calibración y puntos control
Para construir la curva de calibración en seis niveles de concentración por triplicado y los puntos control en cuatro niveles de concentración —muestra control baja, muestra control media, muestra control alta y límite de cuantificación— se adicionaron a 100 µL de plasma libre de fármacos, 50 µL de solución de estándar interno, diferentes volúmenes de solución A o B de lamotrigina según corresponda para alcanzar la concentración deseada en cada punto. El volumen final fue de 1000 µL con metanol.
Todas las muestras se agitaron mecánicamente por 100 segundos y se centrifugaron a 13,400 rpm durante 10 minutos. El sobrenadante obtenido se transfirió a un vial y se leyó en el cromatógrafo utilizado, en este caso un HITACHI® Elite LaChrom acoplado a un detector UV-visible y una columna Symetry C18 150X4.6 mm, tamaño de partícula 5 µm.
Preparación de muestras
Se adicionaron a cada 100 µL de muestra plasmática, 50 µL de solución de estándar interno y 850 µL de metanol. Se agitó mecánicamente por 100 segundos y se centrifugó a 13,400 rpm durante 10 minutos. De esta forma se realizó una extracción líquido-líquido con precipitación de proteínas para liberar el fármaco de la matriz biológica. El sobrenadante se transfirió a un vial y se leyó en el cromatógrafo con las condiciones mencionadas en la preparación de la curva de calibración.
Previo a la validación del método, se debe elaborar un protocolo que incluya la descripción detallada, objetivo, definición y descripción de los parámetros analíticos (características de desempeño) que conformarán la validación de acuerdo con el propósito que se pretenda.10 Asimismo, se deben redactar los criterios y procedimientos para realizarlos, sea el caso de metodologías desarrolladas o metodologías comerciales.
Acorde con la normatividad mexicana, los métodos diseñados específicamente para la determinación cuantitativa de un fármaco o biomolécula en muestras biológicas, aplicados ya sea a pruebas de intercambiabilidad (bioequivalencia) o biodisponibilidad en humanos, deben, además, contar con procedimientos normalizados de operación que aseguren la correcta separación (pre proceso), manejo, traslado, identificación, almacén y disposición final de las muestras involucradas en el método.10
El avance tecnológico tanto instrumental como en el procesamiento y/o preparación de diversas muestras biológicas para medir concentraciones de un fármaco con límites de detección cada vez más reducidos hace que la técnica CLAR sea una de las herramientas preferidas en el desarrollo y validación de métodos analíticos para cuantificar fármacos.11,12
La CLAR requiere de muestras pequeñas y se pueden medir varios fármacos en la misma corrida.11 Esencialmente, es un proceso físico donde las moléculas de una mezcla se dispersan por distribución en dos fases: una estacionaria (columna) y la otra móvil (fase móvil).
El proceso se genera a partir de la adsorción y desorción de los componentes de la muestra con el lecho estacionario y su afinidad con la fase móvil13, pero dependen de la combinación de diversas condiciones cromatográficas involucradas como el equipo cromatográfico, tipo de columna, composición de la fase móvil, temperatura del horno, flujo de la bomba, tipo de detector, volumen de inyección y, dependiendo de la metodología, estándar interno.
En la tabla 1 se encuentran, a modo de ejemplo, las condiciones finales utilizadas para cuantificar lamotrigina en plasma humano con un método de CLAR fase reversa desarrollado en nuestro laboratorio.
| Tabla 1. Condiciones cromatográficas del método | |
|---|---|
| Sistema cromatográfico | Cromatógrafo HITACHI®, Elite LaChrom |
| Software | EZChrom Elite® |
| Columna | Symetry C18 150X4.6 mm, tamaño de partícula 5 μm |
| Temperatura | 37°C |
| Fase móvil | Solución de fosfatos 0.03 M pH 4.5 acetonitrilo: metanol; proporción 70:20:10 v/v/v |
| Velocidad de flujo | 1.2 mL/min |
| Longitud de onda | 210 nm |
| Estándar interno | Metilparabeno |
| Volumen de inyección | 20 μL |
| Metodología final para la determinación de lamotrigina en plasma humano. | |
En concordancia con la literatura internacional, la normatividad mexicana también establece que sólo se procederá a la validación del método una vez que se cuente con las condiciones cromatográficas necesarias para la cuantificación del fármaco en la matriz biológica.
Se establece como mínimo la determinación de4-6,10:
- selectividad,
- curva de calibración,
- límite inferior de cuantificación,
- repetibilidad y reproducibilidad (en términos de precisión y exactitud),
- estabilidad a corto y/o largo plazo,
- cualquier requerimiento extra según la metodología y el objetivo del protocolo.
Los parámetros analíticos (características de desempeño) fueron contemplados en este estudio.
Selectividad
Es el parámetro cuyo objetivo es demostrar que las moléculas endógenas y exógenas asociadas a la matriz biológica no interfieren entre sí en la señal cromatográfica. Se determina mediante la evaluación individual de al menos 6 diferentes unidades de ésta.10
La selectividad para este ejemplo se evalúo tal como lo marca la normatividad mexicana10: ejecutando el blanco (matriz biológica sin analito de interés ni estándar interno); donde se encontró
- la señal típica del frente del disolvente y el control cero (matriz biológica con estándar interno y sin analito);
- la señal típica del frente del disolvente y un pico representativo del estándar interno.
Adicionalmente, se requiere establecer una adecuada resolución (Rs > 1.5) entre los picos obtenidos (que no deben sobreponerse), por lo que se analizó una muestra que contiene ambos (analito y estándar interno) a una concentración de 5 μg/mL y 10μg/mL, respectivamente. El cálculo de la resolución se efectúo con la siguiente operación matemática:
Rs=(t2-t1)/((1/2)(w1+w2))
En esta expresión matemática (t) se refiere al tiempo de retención y (w) a la anchura del pico. En la gráfica 1 se observa el ejemplo de la corrida analítica de lamotrigina y su estándar interno, donde se demuestra que no existe sobreposición entre ellos. El resultado es una resolución mayor a 1.5 entre los picos.
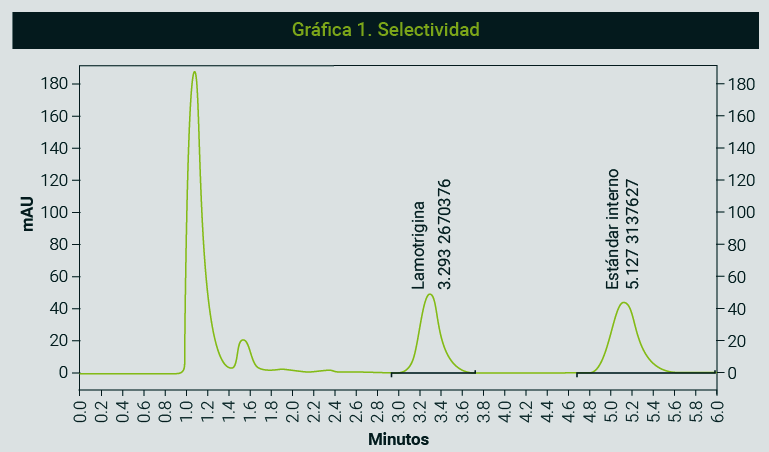
Es importante mencionar que, en el ejemplo descrito, al tratarse de un método con finalidad de monitoreo farmacológico, la evaluación de la selectividad contempló también la evaluación de la no interferencia de posibles fármacos de uso concomitante (topiramato, levetiracetam, oxcarbazepina, paracetamol, naproxeno y diclofenaco) con la respuesta de nuestro fármaco de interés (lamotrigina).13,14
Señales cromatográficas para oxcarbazepina, paracetamol y levetiracetam fueron detectadas con el método propuesto en los 4.2 minutos, 1.65 minutos y 1.15 minutos, respectivamente. Ninguna de ellas interfirió con la señal de lamotrigina. El resto de fármacos evaluados (topiramato, naproxeno y diclofenaco) no mostró señal alguna empleando el método señalado. Por lo tanto, el método es capaz de diferenciar el analito de interés en la matriz biológica de agentes endógenos y exógenos.
Curva de calibración
Se establece conforme a la aplicación cuantitativa que pretenda cada método en específico, es decir, en función de las concentraciones esperadas del analito de interés.
Para este ejemplo, las concentraciones de lamotrigina se eligieron conforme al intervalo terapéutico reportado entre 3 y 14 µg/mL del fármaco.13,14 En la gráfica 2 se observa que, para el ejemplo de la lamotrigina, existe una relación lineal continua y reproducible del tipo: y=mx+b, que se describe entre el rango de concentraciones establecido para el método (0.3 μg/mL, 0.5 μg/mL, 1 μg/mL, 5 μg/mL, 10 μg/mL y 50 μg/mL) y la señal cromatográfica (relación de áreas bajo la curva).
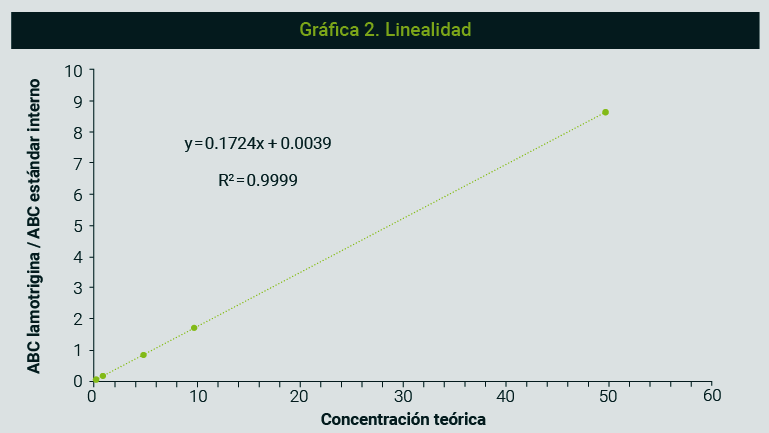
Dentro de este intervalo, el porcentaje de desviación para las concentraciones recuperadas sobre el del valor nominal fue menor al 15% en cada nivel, incluido el límite inferior de cuantificación. Lo anterior cumple satisfactoriamente lo establecido por la normatividad respecto a la curva de calibración, la cual indica que se debe demostrar relación entre la concentración nominal del analito de interés y la respuesta del método en un intervalo de trabajo de seis concentraciones diferentes en función de las respuestas esperadas durante el análisis de las muestras problema.
Las concentraciones no deben incluir la muestra blanco y debe realizarse por triplicado en la misma matriz biológica que las muestras a analizar.10 La curva de calibración es válida si la relación entre la concentración y la respuesta es continua y reproducible en el intervalo de trabajo y si el porcentaje de desviación para la concentración recuperada sobre el del valor nominal es menor al 15% en cada punto, excepto para el límite inferior de cuantificación, en cuyo caso se acepta hasta un 20%.10
Límite inferior de cuantificación (LIC)
Se debe determinar con base en el 5% de la concentración máxima (Cmáx), reportada para el analito de interés, a menos que los objetivos del estudio especifiquen algo diferente, por ejemplo, un muestreo truncado, distribución rápida o alta variabilidad farmacocinética.10
Para que el límite inferior de cuantificación sea validado, se debe llevar a cabo la evaluación por quintuplicado de la concentración nominal más baja y al obtener las concentraciones recuperadas, éstas deben encontrarse dentro del +/-20% de desviación promedio para la exactitud y no exceder el 20% de coeficiente de variación para la precisión.
El LIC definido para la cuantificación de lamotrigina en plasma humano en el ejemplo presentado fue de 0.3 μg/mL, y como puede observarse en la tabla 2, se encuentra dentro de los parámetros establecidos en la normatividad, lo cual indica que la mínima concentración de lamotrigina en la curva propuesta puede ser cuantitativamente determinada con exactitud y precisión aceptables.
| Tabla 2. Límite inferior de cuantificación | |
|---|---|
| Concentración teórica (μg/mL) | Concentración experimental (μg/mL) |
| 0.3 | 0.3061 |
| 0.3 | 0.2953 |
| 0.3 | 0.2838 |
| 0.3 | 0.3082 |
| 0.3 | 0.4130 |
| %CV | 0.0702 |
| %Dprom | 2.3238 |
| Análisis por quintuplicado de la concentración de lamotrigina más baja del rango de trabajo (3 μg/mL) propuesto para su cuantificación. El promedio del porcentaje de desviación (%Dprom) y el porcentaje de coeficiente de variación (%CV) es menor a 20%; por lo tanto, existe exactitud y precisión. | |
Precisión
Se refiere a la concordancia entre resultados individuales al aplicar repetidamente un procedimiento analítico experimental. Este parámetro es referido tanto en literaturas internacionales como en la normatividad mexicana, valorada en términos de repetibilidad y reproducibilidad.4-6,10
En las tablas 3 y 4 se presentan resultados para la cuantificación de lamotrigina, obtenidos al aplicar lo establecido en la normatividad para los parámetros de precisión en términos de repetibilidad y reproducibilidad, respectivamente. Puede observase que el método es preciso de acuerdo con los criterios de aceptación vigentes.
La repetibilidad valora la precisión en semejantes condiciones de operación.
Analizando por quintuplicado en un mismo día, los puntos control bajo, medio y alto, además del LIC, cuyos valores nominales deben empatar con el intervalo de trabajo de la curva validada.
La concentración para cada nivel por interpolación de su respuesta analítica en la curva debe mantener un coeficiente de variación (%CV) ≤ 15% en todos los puntos analizados, excepto para el LIC, el cual permite hasta 20%.10
En tanto, la reproducibilidad (llamada de intralaboratorio por la normatividad mexicana) toma en cuenta la precisión bajo diferentes condiciones que pueden cambiar en el sitio de aplicación: días diferentes, cambio de analista, modificación o cambio de equipos, entre otros.
Los requisitos normativos destacan la valoración de la reproducibilidad del método para diferentes días al aplicar:
- un análisis al menos por quintuplicado similar al de la precisión, con la diferencia de ejecutarlo en tres corridas analíticas diferentes y en al menos dos días,
- el cumplimiento de criterios de aceptación de forma similar que en la repetibilidad del método.10
Exactitud
La gran mayoría de las guías para validación de métodos bioanalíticos por CLAR para la cuantificación de analitos en muestras biológicas describe el desarrollo experimental y criterios de aceptación para el parámetro de exactitud de forma conjunta con los de precisión del método.4,5
En la normatividad mexicana10 no es así, los parámetros se encuentran descritos separadamente; no obstante, la evaluación del parámetro de exactitud indica que se deberá llevar a cabo mediante el uso de datos de repetibilidad y reproducibilidad (lo cual hace una diferencia sólo de forma, pero no de concepto) al calcular la desviación de la concentración obtenida respecto al valor nominal (% de desviación) empleando la siguiente ecuación:
% de desviación=(100)((concentración adicionada-concentración obtenida)/(concentración adicionada))
Los resultados generados para la exactitud del método intra día o en días diferentes para los cuatro puntos control ensayados se presentan en las tabla 3 y 4. De acuerdo con los resultados obtenidos para este parámetro, se puede indicar que el método se considera exacto, ya que el valor promedio del porcentaje de desviación no fue mayor al 15% en ningún punto control analizado, incluido el límite inferior de cuantificación, el cual puede tener un porcentaje de desviación menor o igual a 20%.10
| Tabla 3. Repetibilidad del método | ||||
|---|---|---|---|---|
| Precisión (repetiblidad) | Exactitud | |||
| Concentración teórica (μg/mL) | Concentración experimental promedio (μg/mL) | %CV | %Desviación | |
| LIC | 0.3 | 0.34+/-0.0093 | 2.77 | 12.08 |
| Control bajo | 0.4 | 0.44+/-0.0251 | 5.74 | 9.07 |
| Control medio | 4 | 3.86+/-0.0603 | 1.56 | -4.67 |
| Control alto | 30 | 29.87+/-0.554 | 1.86 | -0.44 |
| Análisis por sextuplicado de los puntos control bajo, medio, alto y del LIC que se encuentran dentro del rango de trabajo en un mismo día. En todos los casos cumple con los criterios de aceptación indicados en la normatividad. | ||||
| Tabla 4. Reproducibilidad del método | ||||
|---|---|---|---|---|
| Precisión (repetiblidad) | Exactitud | |||
| Concentración teórica (μg/mL) | Concentración experimental promedio (μg/mL) | %CV | %Desviación | |
| LIC | 0.3 | 0.32+/-0.0134 | 4.19 | 7.18 |
| Control bajo | 0.4 | 0.43+/-0.0046 | 1.06 | 7.86 |
| Control medio | 4 | 3.90+/-0.037 | 0.95 | -3.58 |
| Control alto | 30 | 29.63+/-0.5152 | 1.74 | -2.25 |
| Análisis por sextuplicado de tres corridas analíticas diferentes y en tres días diferentes de los puntos control bajo, medio, alto y del LIC. Todos los puntos control cumplen los criterios de aceptación normativos. | ||||
Estabilidad
Este parámetro es crucial en la validación, ya que el análisis cuantitativo del fármaco en la muestra biológica no se realiza inmediatamente después de su recolección. Generalmente, las muestras primero se someten a un proceso de separación y el sobrenadante de interés se resguarda para su posterior análisis según el procedimiento establecido.15
Las condiciones para que un analito se mantenga estable dentro de la matriz a analizar depende del manejo, proceso y, sobre todo, del almacenaje acorde con la metodología propuesta. Por ello, las condiciones de almacenamiento deben cubrir todas las situaciones relevantes a las que se enfrentará la muestra en la práctica.
La normatividad mexicana no difiere de la literatura internacional al referir que la estabilidad debe evaluarse por triplicado del análisis del punto control bajo y alto una vez que se preparan y se someten a las diferentes condiciones propuestas, según sea el caso e indicación del protocolo:
- a corto o largo plazo,
- muestra procesada,
- estabilidad en el automuestreador,
- ciclos de congelación-descongelación, entre otros.
Incluso se marca la evaluación de la demostración de la estabilidad de la solución de referencia principal en caso de no utilizarla de forma inmediata.
Se compara la desviación promedio de las concentraciones obtenidas posterior a cada condición de estabilidad contra la concentración nominal inicial, que no debe exceder el +/-15% CV, respecto a la concentración nominal para ser consideradas confiables.10 Para ejemplificar este punto se construyeron las tablas 5 y 6, en las que se presentan los resultados del reto de estabilidad en la cuantificación de lamotrigina en plasma humano tras el resguardo en refrigeración (5+/- 3 °C) de las muestras a corto plazo (1, 2 y 3 días) y a largo plazo (15 y 90 días), respectivamente.
Como se puede observar, el punto control bajo (0.4 μg/mL) y del punto control alto (30 μg/mL) se encuentran dentro del 15% CV permitido. Ello significa que las muestras son estables en condiciones de refrigeración al menos durante 90 días de almacenamiento.
| Tabla 5. Estabilidad | ||||||
|---|---|---|---|---|---|---|
| A corto plazo* | ||||||
| Día 1 | Día 2 | Día 3 | ||||
| Concentración nominal | Concentración experimental promedio (μg/mL) | %CV | Concentración experimental promedio (μg/mL) | %CV | Concentración experimental promedio (μg/mL) | %CV |
| PCB (0.4 μg/mL) | 0.42+/-0.0102 | 2.41 | 0.40+/-0.0043 | 1.07 | 0.37+/-0.0024 | 0.64 |
| PCA (30 μg/mL) | 30.60+/-1.7842 | 5.83 | 29.07+/-0.6087 | 2.09 | 27.55+/-0.2319 | 0.84 |
| A largo plazo** | ||||
|---|---|---|---|---|
| Día 15 | Día 90 | |||
| Concentración nominal | Concentración experimental promedio (μg/mL) | %CV | Concentración experimental promedio (μg/mL) | %CV |
| PCB (0.4 μg/mL) | 0.40+/-0.0085 | 2.12 | 0.39+/-0.0058 | 1.49 |
| PCA (30 μg/mL) | 29.63+/-0.1276 | 0.43 | 29.55+/-0.1403 | 0.47 |
| *Análisis del punto control bajo (PCB) y del punto control alto (PCA) de muestras plasmáticas que contienen lamotrigina almacenadas en refrigeración (5+/-3°C) a corto plazo. Cada dato se presenta como el promedio de tres determinaciones. En ningún caso se obtuvo un%CV mayor a 15% **Análisis del punto control bajo (PCB) y del punto control alto (PCA) de muestras plasmáticas que contienen lamotrigina en condiciones de refrigeración (5+/-3°C) hasta por 90 días. Cada dato se presenta como el promedio de tres determinaciones. Ninguna determinación obtuvo un%CV mayor a 15%. | ||||
Para el campo biofarmacéutico, desarrollar metodologías que puedan cuantificar fármacos en matriz biológica por medio de técnicas cromatográficas abarca varios ámbitos. Obtener información acerca de la biodisponibilidad de los fármacos de interés es uno de ellos.16-18
Algunos estudios de intercambiabilidad comparan la biodisponibilidad de un medicamento de prueba contra uno de referencia; si no hay diferencia estadística entre ellos se dice que los medicamentos son bioequivalentes.18,19
Para ensayos clínicos, vigilar los niveles de concentración de un fármaco en el organismo permite establecer regímenes de dosificación adecuados.20,21 También, emplear métodos analíticos para cuantificar fármacos de ventana terapéutica estrecha tiene como finalidad comprobar que la dosis administrada sea la asociada a seguridad y eficacia, es decir, la llamada monitorización de fármacos.20,21
Cualquiera que sea el objetivo del método validado, la última fase de la validación es su aplicación al análisis real de las muestras. Para el ejemplo que compete en este escrito, se realizó la cuantificación del fármaco en muestras plasmáticas provenientes de pacientes a los cuales se les administró lamotrigina vía oral con la finalidad de evidenciar la aplicación del método en el monitoreo de los niveles séricos del fármaco.
En la gráfica 3 se presentan cursos temporales de concentración plasmática de lamotrigina en función del tiempo que evidencia satisfactoriamente la aplicación de la metodología validada para cuantificar la sustancia de interés en plasma por medio de la técnica CLAR, acorde con los requisitos de la NOM-177-SSA1-2013 (modificación reciente).22 Los resultados sugieren que los niveles plasmáticos de lamotrigina en los pacientes se encuentran en el intervalo terapéutico señalado en la bibliografía.13,14
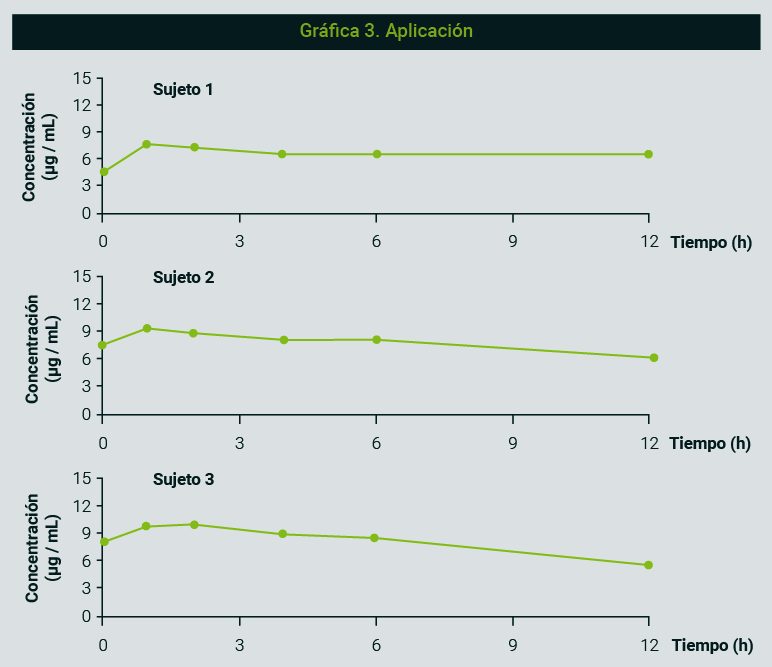
Las guías de validación que existen en México suelen ser complejas y no siempre se aplican a la validación de métodos bioanalíticos.
En este trabajo se enumeraron los requerimientos mínimos y los criterios de aceptación que establece la NOM-177-SSA1-2013 para validar métodos analíticos que cuantifican fármacos en matriz biológica por medio de la técnica CLAR con el objetivo de asegurar que los resultados obtenidos sean confiables y aplicables acorde con el alcance de cada metodología según su protocolo.
Los resultados del método analítico para cuantificar lamotrigina en plasma humano (con la técnica CLAR) desarrollado en nuestro laboratorio demostraron que dicho método es lineal, selectivo, repetible, reproducible, exacto y estable para su aplicación, ya que cumple con los criterios de aceptación que establece la normatividad mexicana. Por lo tanto, los resultados pueden ser aplicados de manera correcta para dar respuesta a problemáticas actuales del sector salud, en este caso, llevar acabo un monitoreo terapéutico adecuado en pacientes que toman lamotrigina de forma regular en su tratamiento.
Agradezco al Consejo Nacional de Humanidades Ciencias y Tecnologías (CONAHCYT) por la beca 828452 otorgada para realizar estudios de Maestría; y al Dr. Gilberto Castañeda Hernández por la asesoría brindada en este proyecto.
Todos los autores declaran no tener conflicto de interés alguno.
| 1. | Andrews J. Validating pharmaceutical systems. Good computer practice in life science manufacturing. Boca Raton: Tailor & Francis, 2005. |
| 2. | Shah VP, Bansal S. Historical perspective on the development and evolution of bioanalytical guidance and technology. Bioanalysis. 2011; 3(8):823-7. |
| 3. | Bansal S, DeStefano A. Key elements of bioanalytical method validation for small molecules. AAPS J. 2007;9(1): E109-E114. |
| 4. | Food and Drug Administration. Bioanalytical method validation guidance for industry. 2018. [Retrieved on July 11th, 2024]. Available from URL: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry |
| 5. | European Medicines Agency. ICH M10 on bioanalytical method validation. Scientific guideline. 2023. [Retrieved on July 11th, 2024]. Available from URL: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation |
| 6. | International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). M10: Bioanalytical method validation and study sample analysis. [Retrieved on July 18th, 2024]. Available from URL: https://database.ich.org/sites/default/files/M10_Guideline_Step4_2022_0524.pdf |
| 7. | Colegio Nacional de Químicos Farmacéuticos Biólogos. Guía de validación de métodos analíticos. 2002 [Consultado 11 de julio, 2024]. Disponible en URL: https://colegioqfb.org.mx/recurso/g/ |
| 8. | Arrieta Sánchez M, Flores Gómez I, Galicia Rosas MR, Huerta Flores L, López González LM. Manual de desarrollo analítico (Laboratorio) SGC-FESZ-QFB-ML19, UNAM. 2020. Disponible en URL: https://www.zaragoza.unam.mx/wp-content/Portal2015/Licenciaturas/qfb/manuales/9Manual_Desarrollo_Analitico.pdf |
| 9. | Secretaría de Salud. FEUM. Farmacopea de los Estados unidos mexicanos. Edición 13. 2021. [Consultado 11 de julio, 2024] Disponible en URL: https://www.farmacopea.org.mx/publicaciones-detalle.php?m=3&pid=12 |
| 10. | Secretaría de Gobernación. Diario Oficial de la Federación. Norma Oficial Mexicana NOM-177-SSA1-2013. [Consultado 11 de julio, 2024].Disponible en URL: https://www.dof.gob.mx/nota_detalle.php?codigo=5298030&fecha=06/05/2013#gsc.tab=0 |
| 11. | Sahu PK, Ramisetti NR, Cecchi T, Swain S, Patro CS, Panda J. An overview of experimental designs in HPLC method development and validation. J Pharm Biomed Anal. 2018;147:590-611. |
| 12. | Bajda L, Amaro MM, Bongiovanni GA. Métodos cromatográficos optimizados para la identificación y cuantificación de terpenos en aceite de Cannabis sativa de uso medicinal [Optimized chromatographic methods for the identification and quantification of terpenes in Cannabis sativa oil for medicinal use]. Rev Fac Cien Med Univ Nac Cordoba. 2023;80(2):99-105. |
| 13. | SEUP. Guía rápida para el tratamiento de las intoxicaciones pediátricas. Lamotrigina. Antiepilépticos. 2024. [Consultado 11 de julio, 2024] Disponible en URL: https://toxseup.org/lamotrigina/ |
| 14. | Aldaza A, Ferriols R, Aumentec D, Calvod MV, Farree MR, García B, et al. Monitorización farmacocinética de antiepilépticos. Farm Hosp. 2011; 35(6):326-339. |
| 15. | van de Merbel N, Savoie N, Yadav M, Ohtsu Y, White J, Riccio M, et al. Stability: Recommendation for best practices and harmonization from the Global Bioanalysis Consortium Harmonization Team. AAPS J. 2014;16(3):392-399. |
| 16. | Zhao C, Zheng N, Yang F, Han SY, Li PP. A validated high-performance liquid chromatography-tandem mass spectrometry method for quantification of gefitinib and its main metabolites in xenograft mouse tumor: Application to a pharmacokinetics study. Biomed Chromatogr. 2019;33(11):e4638. |
| 17. | Perdomo F, Cabrera Fránquiz F, Cabrera J, Serra-Majem L. Influencia del procedimiento culinario sobre la biodisponibilidad del licopeno en el tomate [Influence of cooking procedure on the bioavailability of lycopene in tomatoes]. Nutr Hosp. 2012;27(5):1542-1546. |
| 18. | Saavedra SI, Sasso AJ, Quiñones SL, Saavedra BM, Gaete GL, Boza TI, et al. Estudio de biodisponibilidad relativa entre dos formulaciones orales de micofenolato mofetilo en voluntarios sanos [Relative bioavailability study of two oral formulations of mycophenolate mofetil in healthy volunteers]. Rev Med Chil. 2011;139(7):902-908. |
| 19. | Flores Murrieta FJ, Castañeda Hernández G, Medina Santillán R. Biodisponibilidad y bioequivalencia en los medicamentos genéricos. México: Editorial Asclepius XXI; 2002. |
| 20. | Escobar L. Therapeutic drug monitoring and practical aspects of pharmacokinetics. RMCLC. 2016; 27(5):605-614. [Consultado 11 de julio, 2024]. Disponible en URL: https://www.elsevier.es/es-revista-revista-medica-clinica-las-condes-202-pdf-S0716864016300864 |
| 21. | Kang JS, Lee MH. Overview of therapeutic drug monitoring. Korean J Intern Med. 2009;24(1):1-10 |
| 22. | Secretaría de Gobernación. Diario Oficial de la Federación. MODIFICACIÓN a la Norma Oficial Mexicana NOM-177-SSA1-2013.2023. [Consultado 11 de julio, 2024]. Disponible en URL: https://www.dof.gob.mx/nota_detalle.php?codigo=5702018&fecha=15/09/2023#gsc.tab=0 |
All Rights Reserved® 2019
Latin American Journal of Clinical Sciences and Medical Technology,Publicación contínua • Editor responsable: Gilberto Castañeda Hernández. • Reserva de Derechos al Uso Exclusivo: 04-2019-062013242000-203; ISSN: 2683-2291; ambos otorgados por el Instituto Nacional del Derecho de Autor. • Responsable de la última actualización de este número, Web Master Hunahpú Velázquez Martínez,
Calle Profesor Miguel Serrano #8, Col. Del Valle, Alcaldía Benito Juárez, CP 03100, Ciudad de México, México. Número telefónico: 55 5405 1396 • Fecha de última modificación, 28 de agosto de 2024.
All Rights Reserved® 2019
Publicación contínua • Editor responsable: Gilberto Castañeda Hernández. • Reserva de Derechos al Uso Exclusivo: 04-2019-062013242000-203; ISSN: 2683-2291; ambos otorgados por el Instituto Nacional del Derecho de Autor. • Responsable de la última actualización de este número, Web Master Hunahpú Velázquez Martínez,
Calle Profesor Miguel Serrano #8, Col. Del Valle, Alcaldía Benito Juárez, CP 03100, Ciudad de México, México. Número telefónico: 55 5405 1396 • Fecha de última modificación, 28 de agosto de 2024.